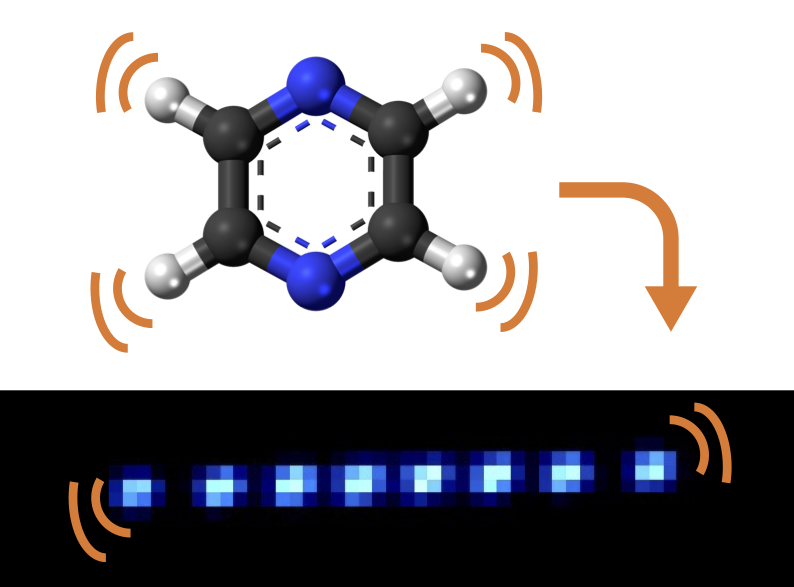
Our quantum-computer simulation of geometric-phase interference35 featured on the cover of Nature Chemistry (Nov 2023).
Simulating chemistry is widely expected to be among the first practically important problems where quantum computers will outperform conventional ones. Faster simulation would accelerate both our understanding of chemistry and the discovery of new functional materials, pharmaceuticals, and other useful molecules.
On conventional computers, predicting chemical properties such as energies or reaction rates rapidly becomes harder for larger molecules. The difficulty of representing quantum effects such as entanglement on classical computers means that the most accurate chemical simulation techniques are restricted to small systems.
Quantum computers could simulate chemistry exponentially faster than classical ones. We have developed a suite of quantum methods that could dramatically accelerate chemical simulations,8 including the simulation of chemical reactions2 and the determination molecular properties.5 We have collaborated with experimentalists to implement these algorithms—including the first quantum-computer calculation of molecular energies6—and we continue to push the limits of quantum technology.
We are currently benchmarking quantum algorithms for simulating chemical dynamics, with the goal of discovering classically intractable reactions that could be explained by quantum computers—if you’re interested, join us!
Analog quantum simulators
Mixed qudit-boson (MQB) simulator for analog quantum simulation of chemical dynamics.31, 34, 35 When implemented in trapped ions, the ion vibrations simulate molecular vibrations. Photo: 8 trapped 173Yb+ ions at the Quantum Control Laboratory, USyd.
Quantum simulators are dedicated devices that can simulate quantum systems without being universal computers. Because they are easier to build than full quantum computers, they promise to be among the first useful quantum technologies.
Our quantum simulators were the first to experimentally demonstrate phenomena that were otherwise too difficult to study in the laboratory, including partially coherent quantum walks,7 topologically protected bound states,9 and environment-assisted quantum transport.17
Recently, we developed mixed qudit-boson (MQB) simulation, a broadly applicable scheme for simulating chemical dynamics on near-term analog quantum simulators31. By taking advantage of bosonic degrees of freedom, we can simulate non-adiabatic processes with a ten-fold reduction in quantum hardware requirements compared to digital quantum computers. We have used this approach on trapped-ion quantum simulators to carry out the first time-dependent quantum simulation of vibronic spectroscopy34 and the first direct observation of geometric phase in any system.35
We are currently furthering our analog simulation platform for chemical processes—stay tuned or come join us!
Christophe H. Valahu*, Vanessa C. Olaya-Agudelo*, Ryan J. MacDonell, Tomas Navickas, Arjun D. Rao, Maverick J. Millican, Juan B. Pérez-Sánchez, Joel Yuen-Zhou, Michael J. Biercuk, Cornelius Hempel, Ting Rei Tan, and Ivan Kassal
Nat. Chem.15, 1503 (2023).
Featured on the front cover of the November 2023 edition
Conical intersections are ubiquitous in chemistry and physics, often governing processes such as light harvesting, vision, photocatalysis and chemical reactivity. They act as funnels between electronic states of molecules, allowing rapid and efficient relaxation during chemical dynamics. In addition, when a reaction path encircles a conical intersection, the molecular wavefunction experiences a geometric phase, which can affect the outcome of the reaction through quantum-mechanical interference. Past experiments have measured indirect signatures of geometric phases in scattering patterns and spectroscopic observables, but there has been no direct observation of the underlying wavepacket interference. Here we experimentally observe geometric-phase interference in the dynamics of a wavepacket travelling around an engineered conical intersection in a programmable trapped-ion quantum simulator. To achieve this, we develop a technique to reconstruct the two-dimensional wavepacket densities of a trapped ion. Experiments agree with the theoretical model, demonstrating the ability of analogue quantum simulators—such as those realized using trapped ions—to accurately describe nuclear quantum effects.
Ryan J. MacDonell*, Tomas Navickas*, Tim F. Wohlers-Reichel, Christophe H. Valahu, Arjun D. Rao, Maverick J. Millican, Michael A. Currington, Michael J. Biercuk, Ting Rei Tan, Cornelius Hempel, and Ivan Kassal
Spectroscopy is one of the most accurate probes of the molecular world. However, predicting molecular spectra accurately is computationally difficult because of the presence of entanglement between electronic and nuclear degrees of freedom. Although quantum computers promise to reduce this computational cost, existing quantum approaches rely on combining signals from individual eigenstates, an approach whose cost grows exponentially with molecule size. Here, we introduce a method for scalable analog quantum simulation of molecular spectroscopy: by performing simulations in the time domain, the number of required measurements depends on the desired spectral range and resolution, not molecular size. Our approach can treat more complicated molecular models than previous ones, requires fewer approximations, and can be extended to open quantum systems with minimal overhead. We present a direct mapping of the underlying problem of time-domain simulation of molecular spectra to the degrees of freedom and control fields available in a trapped-ion quantum simulator. We experimentally demonstrate our algorithm on a trapped-ion device, exploiting both intrinsic electronic and motional degrees of freedom, showing excellent quantitative agreement for a single-mode vibronic photoelectron spectrum of SO2.
Ultrafast chemical reactions are difficult to simulate because they involve entangled, many-body wavefunctions whose computational complexity grows rapidly with molecular size. In photochemistry, the breakdown of the Born-Oppenheimer approximation further complicates the problem by entangling nuclear and electronic degrees of freedom. Here, we show that analog quantum simulators can efficiently simulate molecular dynamics using commonly available bosonic modes to represent molecular vibrations. Our approach can be implemented in any device with a qudit controllably coupled to bosonic oscillators and with quantum hardware resources that scale linearly with molecular size, and offers significant resource savings compared to digital quantum simulation algorithms. Advantages of our approach include a time resolution orders of magnitude better than ultrafast spectroscopy, the ability to simulate large molecules with limited hardware using a Suzuki-Trotter expansion, and the ability to implement realistic system-bath interactions with only one additional interaction per mode. Our approach can be implemented with current technology; e.g., the conical intersection in pyrazine can be simulated using a single trapped ion. Therefore, we expect our method will enable classically intractable chemical dynamics simulations in the near term.
Devon N. Biggerstaff, René Heilmann, Aidan A. Zecevik, Markus Gräfe, Matthew A. Broome, Alessandro Fedrizzi, Stefan Nolte, Alexander Szameit, Andrew G. White, and Ivan Kassal
Transport phenomena on a quantum scale appear in a variety of systems, ranging from photosynthetic complexes to engineered quantum devices. It has been predicted that the efficiency of coherent transport can be enhanced through dynamic interaction between the system and a noisy environment. We report an experimental simulation of environment-assisted coherent transport, using an engineered network of laser-written waveguides, with relative energies and inter-waveguide couplings tailored to yield the desired Hamiltonian. Controllable-strength decoherence is simulated by broadening the bandwidth of the input illumination, yielding a significant increase in transport efficiency relative to the narrowband case. We show integrated optics to be suitable for simulating specific target Hamiltonians as well as open quantum systems with controllable loss and decoherence.
Large-scale quantum computers will require the ability to apply long sequences of entangling gates to many qubits. In a photonic architecture, where single-qubit gates can be performed easily and precisely, the application of consecutive two-qubit entangling gates has been a significant obstacle. Here, we demonstrate a two-qubit photonic quantum processor that implements two consecutive CNOT gates on the same pair of polarisation-encoded qubits. To demonstrate the flexibility of our system, we implement various instances of the quantum algorithm for solving of systems of linear equations.
Takuya Kitagawa*, Matthew A. Broome*, Alessandro Fedrizzi, Mark S. Rudner, Erez Berg, Ivan Kassal, Alán Aspuru-Guzik, Eugene Demler, and Andrew G. White
Topological phases exhibit some of the most striking phenomena in modern physics. Much of the rich behaviour of quantum Hall systems, topological insulators, and topological superconductors can be traced to the existence of robust bound states at interfaces between different topological phases. This robustness has applications in metrology and holds promise for future uses in quantum computing. Engineered quantum systems—notably in photonics, where wavefunctions can be observed directly—provide versatile platforms for creating and probing a variety of topological phases. Here we use photonic quantum walks to observe bound states between systems with different bulk topological properties and demonstrate their robustness to perturbations—a signature of topological protection. Although such bound states are usually discussed for static (time-independent) systems, here we demonstrate their existence in an explicitly time-dependent situation. Moreover, we discover a new phenomenon: a topologically protected pair of bound states unique to periodically driven systems.
The difficulty of simulating quantum systems, well known to quantum chemists, prompted the idea of quantum computation. One can avoid the steep scaling associated with the exact simulation of increasingly large quantum systems on conventional computers, by mapping the quantum system to another, more controllable one. In this review, we discuss to what extent the ideas in quantum computation, now a well-established field, have been applied to chemical problems. We describe algorithms that achieve significant advantages for the electronic-structure problem, the simulation of chemical dynamics, protein folding, and other tasks. Although theory is still ahead of experiment, we outline recent advances that have led to the first chemical calculations on small quantum information processors.
Quantum walks have a host of applications, ranging from quantum computing to the simulation of biological systems. We present an intrinsically stable, deterministic implementation of discrete quantum walks with single photons in space. The number of optical elements required scales linearly with the number of steps. We measure walks with up to 6 steps and explore the quantum-to-classical transition by introducing tunable decoherence. Finally, we also investigate the effect of absorbing boundaries and show that decoherence significantly affects the probability of absorption.
Benjamin P. Lanyon, James D. Whitfield, Geoff G. Gillet, Michael E. Goggin, Marcelo P. Almeida, Ivan Kassal, Jacob D. Biamonte, Masoud Mohseni, Ben J. Powell, Marco Barbieri, Alán Aspuru-Guzik, and Andrew G. White
Nature Chem.2, 106 (2010).
One of 17 “First anniversary highlights” of Nature Chemistry and subject of a “News and Views” (Nature Chemistry2, 76 (2010)).
Exact first-principles calculations of molecular properties are currently intractable because their computational cost grows exponentially with both the number of atoms and basis set size. A solution is to move to a radically different model of computing by building a quantum computer, which is a device that uses quantum systems themselves to store and process data. Here we report the application of the latest photonic quantum computer technology to calculate properties of the smallest molecular system: the hydrogen molecule in a minimal basis. We calculate the complete energy spectrum to 20 bits of precision and discuss how the technique can be expanded to solve large-scale chemical problems that lie beyond the reach of modern supercomputers. These results represent an early practical step toward a powerful tool with a broad range of quantum-chemical applications.
Quantum computers, if available, could substantially accelerate quantum simulations. We extend this result to show that the computation of molecular properties (energy derivatives) could also be sped up using quantum computers. We provide a quantum algorithm for the numerical evaluation of molecular properties, whose time cost is a constant multiple of the time needed to compute the molecular energy, regardless of the size of the system. Molecular properties computed with the proposed approach could also be used for the optimization of molecular geometries or other properties. For that purpose, we discuss the benefits of quantum techniques for Newton’s method and Householder methods. Finally, global minima for the proposed optimizations can be found using the quantum basin hopper algorithm, which offers an additional quadratic reduction in cost over classical multi-start techniques.
While quantum computers are capable of simulating many quantum systems efficiently, the simulation algorithms must begin with the preparation of an appropriate initial state. We present a method for generating physically relevant quantum states on a lattice in real space. In particular, the present algorithm is able to prepare general pure and mixed many-particle states of any number of particles. It relies on a procedure for converting from a second-quantized state to its first-quantized counterpart. The algorithm is efficient in that it operates in time that is polynomial in all the essential descriptors of the system, the number of particles, the resolution of the lattice, and the inverse of the maximum final error. This scaling holds under the assumption that the wave function to be prepared is bounded or its indefinite integral is known and that the Fock operator of the system is efficiently simulatable.
The computational cost of exact methods for quantum simulation using classical computers grows exponentially with system size. As a consequence, these techniques can be applied only to small systems. By contrast, we demonstrate that quantum computers could exactly simulate chemical reactions in polynomial time. Our algorithm uses the split-operator approach and explicitly simulates all electron-nuclear and interelectronic interactions in quadratic time. Surprisingly, this treatment is not only more accurate than the Born–Oppenheimer approximation but faster and more efficient as well, for all reactions with more than about four atoms. This is the case even though the entire electronic wave function is propagated on a grid with appropriately short time steps. Although the preparation and measurement of arbitrary states on a quantum computer is inefficient, here we demonstrate how to prepare states of chemical interest efficiently. We also show how to efficiently obtain chemically relevant observables, such as state-to-state transition probabilities and thermal reaction rates. Quantum computers using these techniques could outperform current classical computers with 100 qubits.