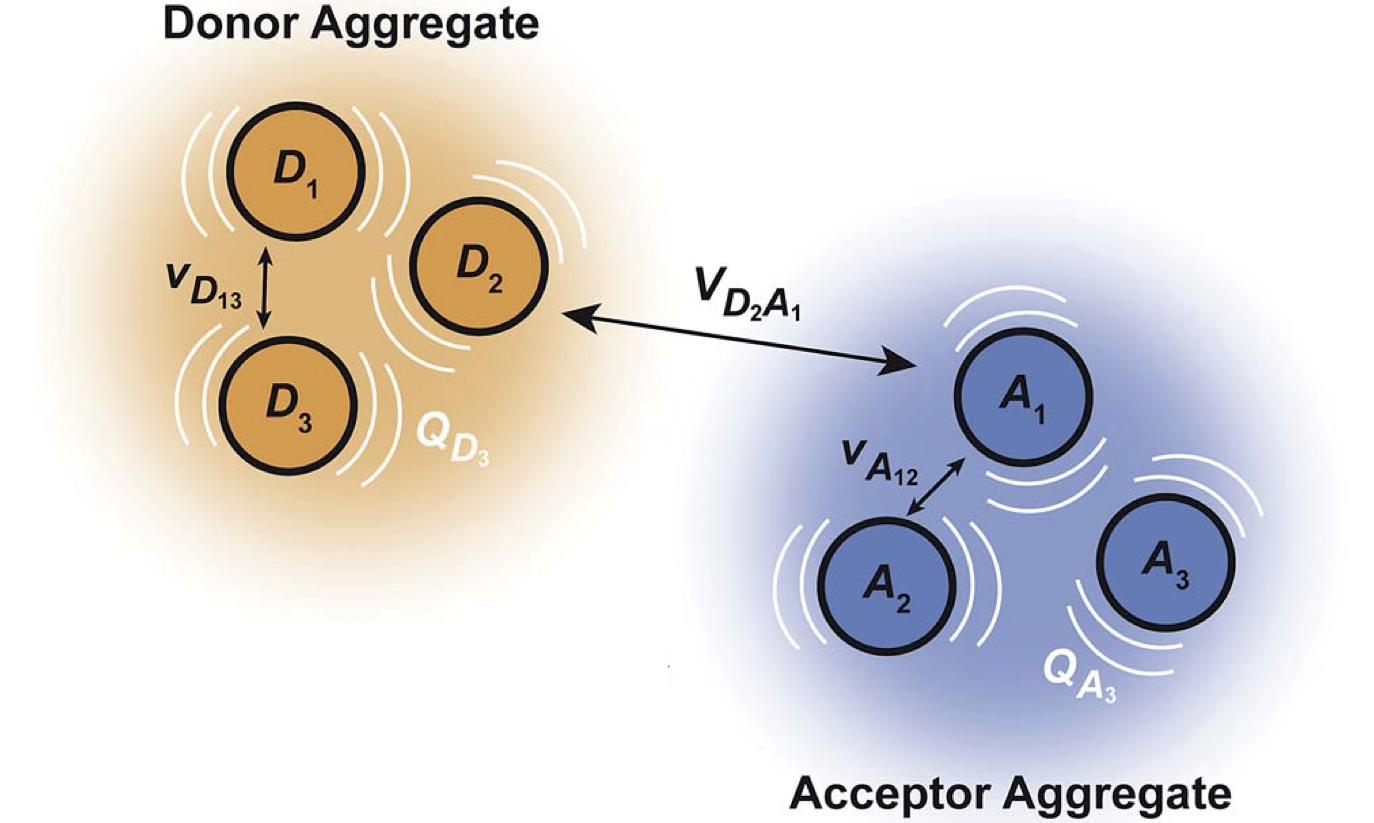
Generalised Marcus theory23 describes the transfer of a charge delocalised over a donor aggregate to an acceptor aggregate.
The movement of electrons or excitations between molecules is perhaps the most fundamental chemical process, one that underpins a vast range of biological and technological functions. We study how charges and excitons move through disordered molecular assemblies such as photosynthetic complexes or organic semiconductors (see below). These materials challenge existing theoretical treatments because they do not fall into any of the well-understood regimes, straddling both order and disorder, both classical and quantum transport.
A recent highlight is our development of generalised Marcus theory,23 a simple approach that allows the transfer of delocalised charges between two molecular aggregates to be described in terms of the properties of the component molecules, offering clear intuition about the role of delocalisation in charge transport.
Organic solar cells
Image: Heliatek
We aim to understand the fundamental physics of organic solar cells, which combine the flexibility and low weight of plastics with the semiconducting properties of conventional photovoltaics. They do not require toxic or rare elements, and promise the fastest energy payback time of any photovoltaic technology. However, many processes in these materials are incompletely understood, making it difficult to formulate precise design principles for better devices.
Most significantly, it remains unclear exactly how the electrons and holes in organic solar cells overcome their electrostatic attraction, sometimes with nearly 100% efficiency. We have shown that entropy and disorder play a crucial role, often being sufficient to overcome the Coulomb barrier.20
To understanding the fundamental photophysics in organic solar cells and facilitate the rational design of better materials, we often collaborate closely with experimentalists. Highlights of these collaborations include:
describing the spectral dependence of the internal quantum efficiency without recourse to hot excitons or other exotic processes13
demonstrating the controlling role of slower charge carriers in the efficiency of charge separation18
developing a new way for measuring energetic disorder in organic semiconductors from their photogeneration efficiency27
Quantum effects in organic semiconductors
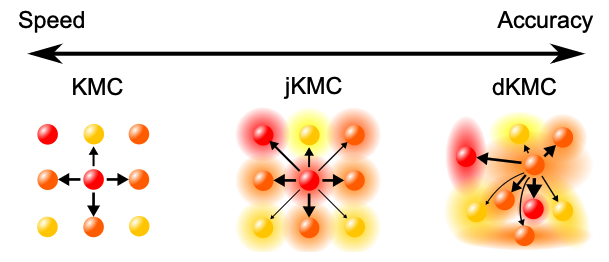
Our delocalised kinetic Monte Carlo (dKMC) describes delocalised transport in disordered materials.30, 33, 37, 40 Jumping kinetic Monte Carlo (jKMC) is a simplified scheme that approaches dKMC in accuracy, with similar computational cost as ordinary KMC.36, 38
We have shown that, just like in photosynthesis, there is a subtle relationship between quantum effects and the separation of charge carriers at organic heterojunctions.24
We have developed delocalised kinetic Monte Carlo (dKMC), a theory of the quantum dynamics of delocalised carriers and excitons in organic semiconductors. dKMC is the first fully quantum treatment that includes the essential ingredients of disorder, delocalisation, and polaron formation in three dimensions and on mesoscopic time and length scales.30, 33, 37, 40 dKMC shows that a little delocalisation goes a long way in organic semiconductors, with realistic delocalisation over only a few molecules able to:
increase charge mobilities by an order of magnitude30
increase exciton mobilities by two orders of magnitude37
increase charge-separation by a factor of five33
increase exciton-dissociation efficiencies by a factor of five40
Our Julia implementation of dKMC is available for download from Github.
We have also developed jKMC, a simplification of dKMC that retains its accuracy but is simple enough to incorporate into any existing kinetic Monte Carlo simulation, for both mobility36 and charge-separation38 calculations.
We are extending our techniques to incorporate quantum effects into other fundamental processes in organic electronics and into device-level simulations—stay tuned or come join us!
Organic photovoltaics (OPVs) are promising candidates for solar-energy conversion, with device efficiencies continuing to increase. However, the precise mechanism of how charges separate in OPVs is not well understood because low dielectric constants produce a strong attraction between the charges, which they must overcome to separate. Separation has been thought to require energetic offsets at donor–acceptor interfaces, but recent materials have enabled efficient charge generation with small offsets, or with none at all in neat materials. Here, we extend delocalised kinetic Monte Carlo (dKMC) to develop a three-dimensional model of charge generation that includes disorder, delocalisation, and polaron formation in every step from photoexcitation to charge separation. Our simulations show that delocalisation dramatically increases charge-generation efficiency, partly by enabling excitons to dissociate in the bulk. Therefore, charge generation can be efficient even in devices with little to no energetic offset, including neat materials. Our findings demonstrate that the underlying quantum-mechanical effect that improves the charge-separation kinetics is faster and longer-distance hops between delocalised states, mediated by hybridised states of exciton and charge-transfer character.
Accurate computational screening of candidate materials promises to accelerate the discovery of higher-efficiency organic photovoltaics (OPVs). However, modelling charge separation in OPVs is challenging because accurate models must include disorder, polaron formation, and charge delocal- isation. Delocalised kinetic Monte Carlo (dKMC) includes these three essential ingredients, but it suffers from high computational cost. Recently, we developed jumping kinetic Monte Carlo (jKMC), a computationally cheap and accurate model of delocalised charge transport that models transport over a lattice of identical, spherical polarons. Here, we extend jKMC to describe the separation of a charge-transfer state, showing that this simplified approach can reproduce the considerable improvements in charge-separation efficiencies caused by delocalisation and first seen in dKMC. The low computational cost and simplicity of jKMC allows it to be applied to parameter regimes intractable by dKMC, and ensures jKMC can be easily incorporated into any existing KMC model.
Large exciton diffusion lengths generally improve the performance of organic semiconductor devices, because they enable energy to be transported farther during the exciton lifetime. However, the physics of exciton motion in disordered organic materials is not fully understood, and modeling the transport of quantum-mechanically delocalized excitons in disordered organic semiconductors is a computational challenge. Here, we describe delocalized kinetic Monte Carlo (dKMC), the first model of three-dimensional exciton transport in organic semiconductors that includes delocalization, disorder, and polaron formation. We find that delocalization can dramatically increase exciton transport; for example, delocalization across less than two molecules in each direction can increase the exciton diffusion coefficient by over an order of magnitude. The mechanism for the enhancement is 2-fold: delocalization enables excitons to hop both more frequently and further in each hop. We also quantify the effect of transient delocalization (short-lived periods where excitons become highly delocalized) and show that it depends strongly upon the disorder and transition dipole moments.
Developing devices using disordered organic semiconductors requires accurate and practical models of charge transport. In these materials, charge transport occurs through partially delocalized states in an intermediate regime between localized hopping and delocalized band conduction. Partial delocalization can increase mobilities by orders of magnitude compared to those with conventional hopping, making it important for the design of materials and devices. Although delocalization, disorder, and polaron formation can be described using delocalized kinetic Monte Carlo (dKMC), it is a computationally expensive method. Here, we develop jumping kinetic Monte Carlo (jKMC), a model that approaches the accuracy of dKMC for modest amounts of delocalization (such as those found in disordered organic semiconductors), with a computational cost comparable to that of conventional hopping. jKMC achieves its computational performance by modeling conduction using identical spherical polarons, yielding a simple delocalization correction to the Marcus hopping rate that allows polarons to jump over their nearest neighbors. jKMC can be used in regimes of partial delocalization inaccessible to dKMC to show that modest delocalization can increase mobilities by as much as 2 orders of magnitude.
In organic photovoltaics, charges can separate efficiently even if their Coulomb attraction is an order of magnitude greater than the available thermal energy. Delocalization has been suggested to explain this fact, because it could increase the initial separation of charges in the charge-transfer (CT) state, reducing their attraction. However, understanding the mechanism requires a kinetic model of delocalized charge separation, which has proven difficult because it involves tracking the correlated quantum-mechanical motion of the electron and the hole in large simulation boxes required for disordered materials. Here, we report the first three-dimensional simulations of charge-separation dynamics in the presence of disorder, delocalization, and polaron formation, finding that even slight delocalization, across less than two molecules, can substantially enhance the charge-separation efficiency, even starting with thermalized CT states. Delocalization does not enhance efficiency by reducing the Coulomb attraction; instead, the enhancement is a kinetic effect produced by the increased overlap of electronic states.
Charge transport is well understood in both highly ordered materials (band conduction) or highly disordered ones (hopping conduction). In moderately disordered materials—including many organic semiconductors—the approximations valid in either extreme break down, making it difficult to accurately model the conduction. In particular, describing wavefunction delocalisation requires a quantum treatment, which is difficult in disordered materials that lack periodicity. Here, we present the first three-dimensional model of partially delocalised charge and exciton transport in materials in the intermediate disorder regime. Our approach is based on polaron-transformed Redfield theory, but overcomes several computational roadblocks by mapping the quantum-mechanical techniques onto kinetic Monte Carlo. Our theory, delocalised kinetic Monte Carlo (dKMC), shows that the fundamental physics of transport in moderately disordered materials is that of charges hopping between partially delocalised electronic states. Our results reveal why standard kinetic Monte Carlo can dramatically underestimate mobilities even in disordered organic semiconductors, where even a little delocalisation can substantially enhance mobilities, as well as showing that three-dimensional calculations capture important delocalisation effects neglected in lower-dimensional approximations.
Quantifying energetic disorder in organic semiconductors continues to attract attention because of its significant impact on the transport physics of these technologically important materials. Here, we show that the energetic disorder of organic semiconductors can be determined from the relationship between the internal quantum efficiency of charge generation and the frequency of the incident light. Our results for a number of materials suggest that energetic disorder in organic semiconductors could be greater than previously reported, and we advance ideas as to why this may be the case.
The dynamics of exciton quenching are critical to the operational performance of organic optoelectronic devices, but their measurement and elucidation remain ongoing challenges. Here, we present a method for quantifying small photoluminescence quenching efficiencies of organic semiconductors under steady-state conditions. Exciton quenching efficiencies of three different organic semiconductors, PC70BM, P3HT, and PCDTBT, are measured at different bulk quencher densities under continuous low-irradiance illumination. By implementing a steady-state bulk-quenching model, we determine exciton diffusion lengths for the studied materials. At low quencher densities we find that a secondary quenching mechanism is in effect, which is responsible for approximately 20% of the total quenched excitons. This quenching mechanism is observed in all three studied materials and exhibits quenching volumes on the order of several thousand cubic nanometers. The exact origin of this quenching process is not clear, but it may be indicative of delocalized excitons being quenched prior to thermalization.
Because of the low dielectric constant, charges in organic solar cells must overcome a strong Coulomb attraction in order to separate. It has been widely argued that intermolecular delocalization would assist charge separation by increasing the effective initial electron–hole separation in a charge-transfer state, thus decreasing their barrier to separation. Here we show that this is not the case: including more than a small amount of delocalization in models of organic solar cells leads to an increase in the free-energy barrier to charge separation. Therefore, if delocalization were to improve the charge separation efficiency, it would have to do so through nonequilibrium kinetic effects that are not captured by a thermodynamic treatment of the barrier height.
Although Marcus theory is widely used to describe charge transfer in molecular systems, in its usual form it is restricted to transfer from one molecule to another. If a charge is delocalised across multiple donor molecules, this approach requires us to treat the entire donor aggregate as a unified supermolecule, leading to potentially expensive quantum-chemical calculations and making it more difficult to understand how the aggregate components contribute to the overall transfer. Here, we show that it is possible to describe charge transfer between groups of molecules in terms of the properties of the constituent molecules and couplings between them, obviating the need for expensive supermolecular calculations. We use the resulting theory to show that charge delocalisation between molecules in either the donor or acceptor aggregates can enhance the rate of charge transfer through a process we call supertransfer (or suppress it through subtransfer). The rate can also be enhanced above what is possible with a single donor and a single acceptor by judiciously tuning energy levels and reorganisation energies. We also describe bridge-mediated charge transfer between delocalised molecular aggregates. The equations of generalised Marcus theory are in closed form, providing qualitative insight into the impact of delocalisation on charge dynamics in molecular systems.
In this Letter, we study the role of the donor:acceptor interface nanostructure upon charge separation and recombination in organic photovoltaic devices and blend films, using mixtures of PBTTT and two different fullerene derivatives (PC70BM and ICTA) as models for intercalated and nonintercalated morphologies, respectively. Thermodynamic simulations show that while the completely intercalated system exhibits a large free-energy barrier for charge separation, this barrier is significantly lower in the nonintercalated system and almost vanishes when energetic disorder is included in the model. Despite these differences, both femtosecond-resolved transient absorption spectroscopy (TAS) and time-delayed collection field (TDCF) exhibit extensive first-order losses in both systems, suggesting that geminate pairs are the primary product of photoexcitation. In contrast, the system that comprises a combination of fully intercalated polymer:fullerene areas and fullerene-aggregated domains (1:4 PBTTT:PC70BM) is the only one that shows slow, second-order recombination of free charges, resulting in devices with an overall higher short-circuit current and fill factor. This study therefore provides a novel consideration of the role of the interfacial nanostructure and the nature of bound charges and their impact upon charge generation and recombination.
The origin of photocurrent losses in the power-generating regime of organic solar cells (OSCs) remains a controversial topic, although recent literature suggests that the competition between bimolecular recombination and charge extraction determines the bias dependence of the photocurrent. Here the steady-state recombination dynamics is studied in bulk-heterojunction OSCs with different hole mobilities from short-circuit to maximum power point. It is shown that in this regime, in contrast to previous transient extracted charge and absorption spectroscopy studies, first-order recombination outweighs bimolecular recombination of photogenerated charge carriers. This study demonstrates that the first-order losses increase with decreasing slower carrier mobility, and attributes them to either mobilization of charges trapped at the donor:acceptor interface through the Poole–Frenkel effect, and/or recombination of photogenerated and injected charges. The dependence of both first-order and higher-order losses on the slower carrier mobility explains why the field dependence of OSC efficiencies has historically been attributed to charge-extraction losses.
Although organic heterojunctions can separate charges with near-unity efficiency and on a sub-picosecond timescale, the full details of the charge-separation process remain unclear. In typical models, the Coulomb binding between the electron and the hole can exceed the thermal energy kT by an order of magnitude, making it impossible for the charges to separate before recombining. Here, we consider the entropic contribution to charge separation in the presence of disorder and find that even modest amounts of disorder have a decisive effect, reducing the charge-separation barrier to about kT or eliminating it altogether. Therefore, the charges are usually not thermodynamically bound at all and could separate spontaneously if the kinetics otherwise allowed it. Our conclusion holds despite the worst-case assumption of localised, thermalised carriers, and is only strengthened if mechanisms like delocalisation or 'hot' states are also present.
Blends of electron-donating and -accepting organic semiconductors are widely used as photoactive materials in next-generation solar cells and photodetectors. The yield of free charges in these systems is often determined by the separation of interfacial electron–hole pairs, which is expected to depend on the ability of the faster carrier to escape the Coulomb potential. Here we show, by measuring geminate and non-geminate losses and key transport parameters in a series of bulk-heterojunction solar cells, that the charge-generation yield increases with increasing slower carrier mobility. This is in direct contrast with the well-established Braun model where the dissociation rate is proportional to the mobility sum, and recent models that underscore the importance of fullerene aggregation for coherent electron propagation. The behaviour is attributed to the restriction of opposite charges to different phases, and to an entropic contribution that favours the joint separation of both charge carriers.
The conventional picture of photocurrent generation in organic solar cells involves photoexcitation of the electron donor, followed by electron transfer to the acceptor via an interfacial charge-transfer state (Channel I). It has been shown that the mirror-image process of acceptor photoexcitation leading to hole transfer to the donor is also an efficient means to generate photocurrent (Channel II). The donor and acceptor components may have overlapping or distinct absorption characteristics. Hence, different excitation wavelengths may preferentially activate one channel or the other, or indeed both. As such, the internal quantum efficiency (IQE) of the solar cell may likewise depend on the excitation wavelength. We show that several model high-efficiency organic solar cell blends, notably PCDTBT:PC70BM and PCPDTBT:PC60/70BM, exhibit flat IQEs across the visible spectrum, suggesting that charge generation is occurring either via a dominant single channel or via both channels but with comparable efficiencies. In contrast, blends of the narrow optical gap copolymer DPP-DTT with PC70BM show two distinct spectrally flat regions in their IQEs, consistent with the two channels operating at different efficiencies. The observed energy dependence of the IQE can be successfully modeled as two parallel photodiodes, each with its own energetics and exciton dynamics but both having the same extraction efficiency. Hence, an excitation-energy dependence of the IQE in this case can be explained as the interplay between two photocurrent-generating channels, without recourse to hot excitons or other exotic processes.